DanaMed Pathfinder biocompatible ACL surgical device, produced with direct metal laser sintering in Inconel 718: an example of a 3D-printed medical device. (Image courtesy Stratasys Direct Manufacturing)


Latest News
January 22, 2018
The U.S. Food and Drug Administration (FDA) recently announced the publication of a 31-page set of guidelines for manufacturers producing medical products via 3D printing/additive manufacturing (AM). Although this information is presented as non-binding recommendations, the agency says it is the first in the world to provide such a comprehensive regulatory framework.
 DanaMed Pathfinder biocompatible ACL surgical device, produced with direct metal laser sintering in Inconel 718: an example of a 3D-printed medical device. Image courtesy of Stratasys Direct Manufacturing.
DanaMed Pathfinder biocompatible ACL surgical device, produced with direct metal laser sintering in Inconel 718: an example of a 3D-printed medical device. Image courtesy of Stratasys Direct Manufacturing.Titled “Technical Considerations for Additively Manufactured Medical Devices: Guidance for Industry and Food and Drug Administration Staff,” the document addresses design, manufacturing and testing considerations of devices only; other work in progress will recommend policies specific to AM of drugs and human tissue. (One example of the latter is the Emerging Technology Program run by the FDA’s Center for Drug Evaluation and Research (CDER). Under this program pharmaceutical companies can connect with the FDA early in their research efforts, which can include use of 3D printing to manufacture drugs.)
A Starting Point for Manufacturers
The FDA, an agency of the U.S. Department of Health and Human Services, is chartered with ensuring the safety, effectiveness and security (among other things) of human and veterinary drugs, vaccines and other biological products for human use, as well as of medical devices.To begin addressing these concerns within the emerging AM field, the FDA reviewed more than 100 devices currently on the market that were manufactured on 3D printers. These ranged from patient-specific knee replacements and cranial implants to medical instruments and surgical guides which may or may not be patient-specific.
This work has been underway for quite some time, beginning with a public workshop held in October 2014, titled, “Additive Manufacturing of Medical Devices: An Interactive Discussion on the Technical Considerations of 3D Printing.” At that meeting, medical device manufacturers, AM companies and academia tackled five broad themes:
- materials,
- design/printing/post-printing validation,
- printing characteristics and parameters,
- physical/mechanical assessment of final devices, and
- biological consideration of final devices (including cleaning, sterility and biocompatibility).
Feedback from that workshop led to publication of a draft version of this document being issued in 2016.
Although designers and manufacturers with years of experience in AM may find some of the document’s information just confirmation of well-known factors in the full 3D-print process (regardless of exact technology), they will also find that this group has compiled a knowledgeable checklist of many process details. The report’s authors note, for example: “The innovative potential of AM may introduce variability into the manufacturing process that would not be present when using other manufacturing techniques,” and “In a powder bed fusion machine, the ratio of reused to virgin powder can affect melting properties, which affects the energy needed to create consistent bonding between layers, which in turn affects final mechanical properties.”
A particular aspect addressed for patient-matched devices (PMD) is the number of factors involved in building parts based on imaging data. One such concern is simply the passage of time, as explained: “When the device is intended to match a patient’s anatomy, and that anatomy can change over time (e.g., with disease progression), the time that can elapse between when the patient is imaged and when the final device is used may need to be reflected in the expiration date of the device.” It then cites information to be included on final-product labeling.
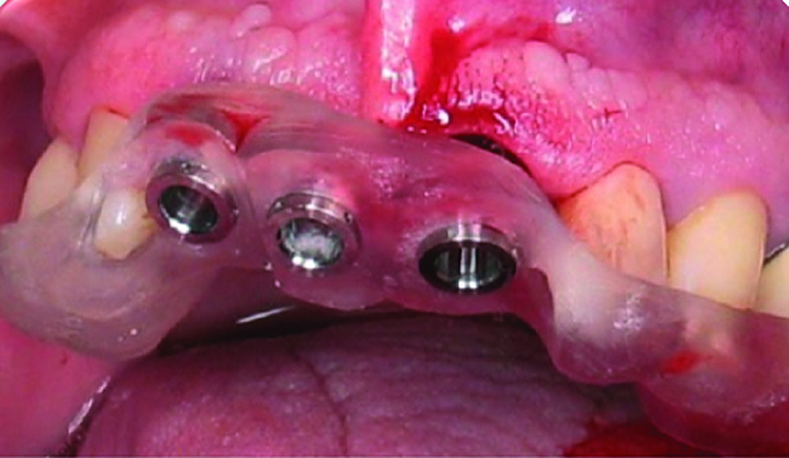 Dental laboratory Protaico uses an Objet Eden260TM 3D Printer to create surgical guides that assist with pre-surgical planning and provide interoperative positioning verification. Image courtesy of Protaico.
Dental laboratory Protaico uses an Objet Eden260TM 3D Printer to create surgical guides that assist with pre-surgical planning and provide interoperative positioning verification. Image courtesy of Protaico.Planning and Documenting the Full AM Process
Other AM-specific topics include the possibility of errors introduced in file-type conversions of data (from imaging to CAD work to printer set-ups), the need for maintaining secure patient data, establishing build-volume control limits for repeatability, and documenting all post-processing steps (i.e., removing manufacturing residues from the device, heat treatments of the device to relieve residual stress, and final machining.)And, since the design and materials involved with any kind of medical devices or implants is subject to improvements, the guidelines recommend: “Manufacturers should rely on existing FDA Guidance for their regulatory pathway when considering a change to a previously cleared or approved device that uses AM.”
Lastly, a key theme throughout the document is the need to establish an in-depth quality system. At a minimum, this should include software workflow, material controls, process validation, material clean-up and sterilization, and mechanical inspection and testing.
Evolving Recommendations
“Today we are issuing new guidance to help advise device manufacturers on technical aspects of 3D printing that clarifies what the FDA recommends manufacturers include on submissions for 3D-printed medical devices,” states FDA commissioner Scott Gottlieb, M.D. in the press release accompanying the new document. “These steps are part of our broader effort to help ensure our regulatory framework is properly matched to the unique attributes of the new technologies we’re being asked to review.”All this information will be useful for manufacturers planning product submissions for FDA approval, and will improve the position of the U.S. in the worldwide AM medical-device marketplace. The FDA encourages interested manufacturers to work closely with the agency starting early in the design cycle.
Subscribe to our FREE magazine, FREE email newsletters or both!
Latest News
About the Author
Pamela Waterman worked as Digital Engineering’s contributing editor for two decades. Contact her via .(JavaScript must be enabled to view this email address).
Follow DE